Difference: Protocols16SSequencing (1 vs. 9)
Revision 92023-09-19 - ElizabethManriquez
16S rRNA Sequencing to Identify Unknown MicrobesEver wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?Overview
PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
It is generally preferred to isolate genomic DNA from your culture before doing the 16s RNA PCR (using the Invitrogen PureLink kit, for example). This results in a cleaner PCR product and generally a better sequencing result. After DNA is isolated, use a Qubit to find the concentration of DNA. The desired amount of gDNA template for PCR is 0.1 ng/ul (final template concentration) so for a 30 ul PCR reaction use 3 ng of DNA. The PCR cycle is as follows:
After PCR, the products need to be run on an agarose gel to confirm correct product (around 1kb). A 1.5% or 2% gel works the best for this. If the bands are clean, then you can do a PCR cleanup spin column and submit the DNA for two different sequencing reactions (one with each of your PCR primers). If the bands are not clean, you may be able to excise the band and The bands should be clean to excise the correct bands from the gel. | |||||||||||||||||
| Changed: | |||||||||||||||||
| < < | After cutting the bands out of gel clean using Zymo gel extraction kit or equivalent. To prepare DNA for sequencing follow the instructions found here: http://www.icmb.utexas.edu/core/DNA/DNA_Sequencing/FAQ-Sequencing.htm | ||||||||||||||||
| > > | After cutting the bands out of gel, clean using Zymo gel extraction kit or equivalent. Proceed to send for Sanger Sequencing. | ||||||||||||||||
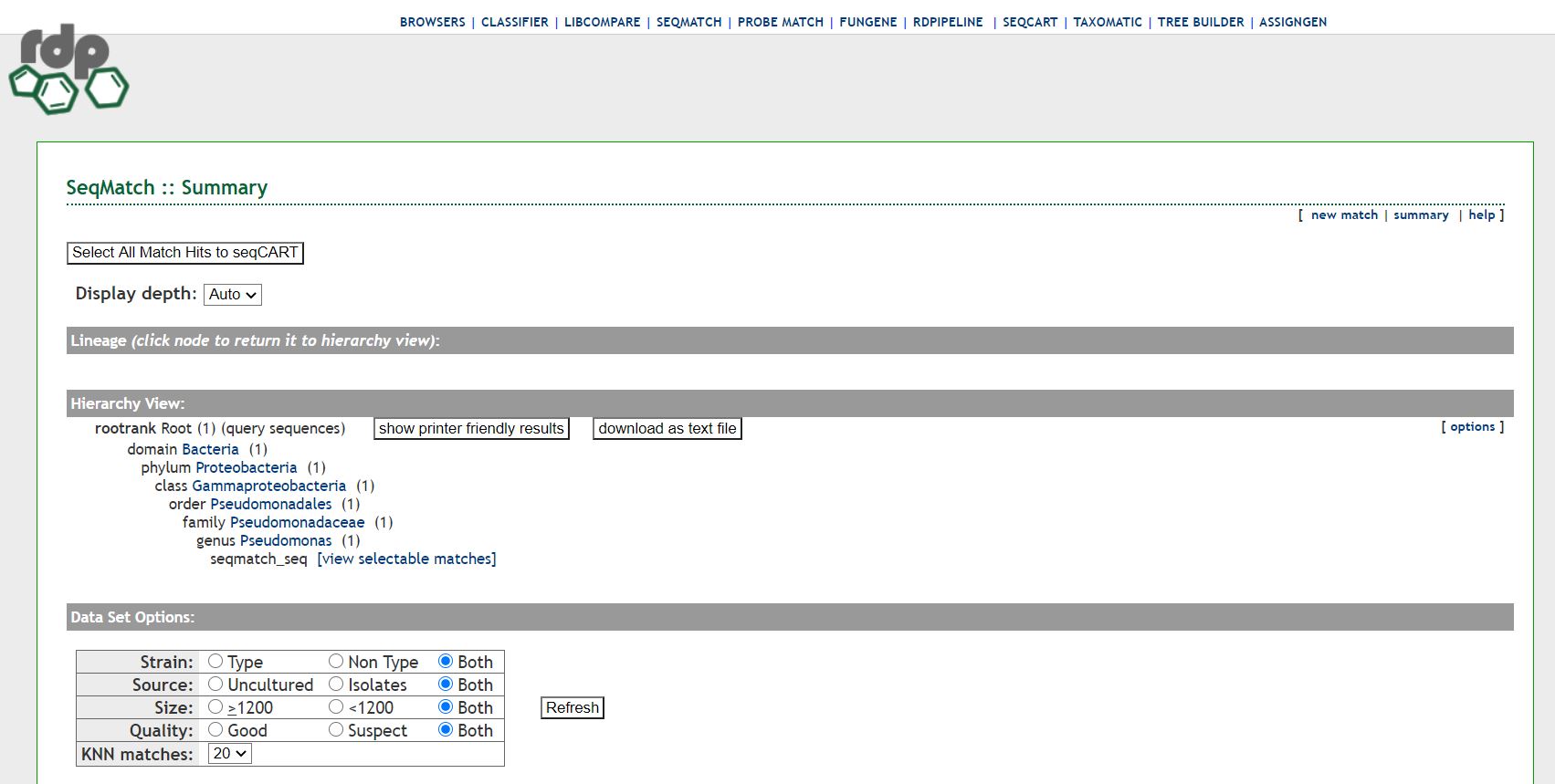
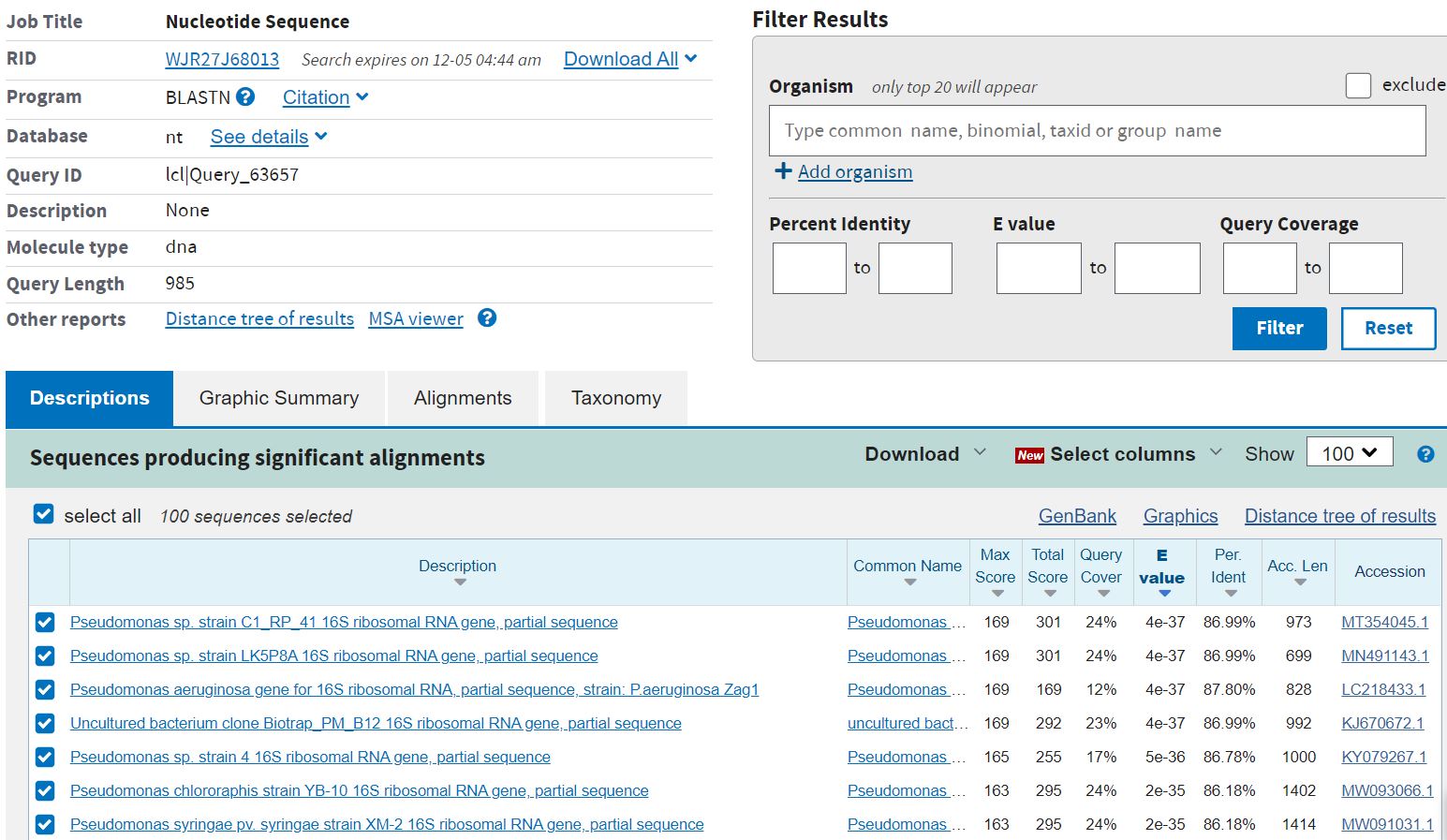
AnalysisUse one of these tools to assign your species:
References1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003).
| |||||||||||||||||
Revision 82020-12-03 - ElizabethRobinson
16S rRNA Sequencing to Identify Unknown MicrobesEver wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?Overview
PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
It is generally preferred to isolate genomic DNA from your culture before doing the 16s RNA PCR (using the Invitrogen PureLink kit, for example). This results in a cleaner PCR product and generally a better sequencing result. After DNA is isolated, use a Qubit to find the concentration of DNA. The desired amount of gDNA template for PCR is 0.1 ng/ul (final template concentration) so for a 30 ul PCR reaction use 3 ng of DNA. The PCR cycle is as follows:
After PCR, the products need to be run on an agarose gel to confirm correct product (around 1kb). A 1.5% or 2% gel works the best for this. If the bands are clean, then you can do a PCR cleanup spin column and submit the DNA for two different sequencing reactions (one with each of your PCR primers). If the bands are not clean, you may be able to excise the band and The bands should be clean to excise the correct bands from the gel. After cutting the bands out of gel clean using Zymo gel extraction kit or equivalent. To prepare DNA for sequencing follow the instructions found here: http://www.icmb.utexas.edu/core/DNA/DNA_Sequencing/FAQ-Sequencing.htm AnalysisUse one of these tools to assign your species:
| |||||||||||||||||
| Added: | |||||||||||||||||
| > > |
| ||||||||||||||||
| |||||||||||||||||
| Added: | |||||||||||||||||
| > > |
| ||||||||||||||||
References1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003). | |||||||||||||||||
| Added: | |||||||||||||||||
| > > |
| ||||||||||||||||
Revision 72016-07-07 - JeffreyBarrick
16S rRNA Sequencing to Identify Unknown MicrobesEver wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?Overview
PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
It is generally preferred to isolate genomic DNA from your culture before doing the 16s RNA PCR (using the Invitrogen PureLink kit, for example). This results in a cleaner PCR product and generally a better sequencing result. After DNA is isolated, use a Qubit to find the concentration of DNA. The desired amount of gDNA template for PCR is 0.1 ng/ul (final template concentration) so for a 30 ul PCR reaction use 3 ng of DNA. The PCR cycle is as follows:
After PCR, the products need to be run on an agarose gel to confirm correct product (around 1kb). A 1.5% or 2% gel works the best for this. If the bands are clean, then you can do a PCR cleanup spin column and submit the DNA for two different sequencing reactions (one with each of your PCR primers). If the bands are not clean, you may be able to excise the band and The bands should be clean to excise the correct bands from the gel. After cutting the bands out of gel clean using Zymo gel extraction kit or equivalent. To prepare DNA for sequencing follow the instructions found here: http://www.icmb.utexas.edu/core/DNA/DNA_Sequencing/FAQ-Sequencing.htm Analysis | |||||||||||||||||
| Changed: | |||||||||||||||||
| < < | Use the Classifer tool | ||||||||||||||||
| > > | Use one of these tools to assign your species: | ||||||||||||||||
| Added: | |||||||||||||||||
| > > |
| ||||||||||||||||
References1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003). | |||||||||||||||||
Revision 62016-05-10 - JeffreyBarrick
16S rRNA Sequencing to Identify Unknown MicrobesEver wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?Overview
PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
| |||||||||
| Changed: | |||||||||
| < < | It is generally preferred to isolate the DNA from your culture before doing the 16s RNA PCR. This results in a cleaner PCR product and generally a better sequencing result. | ||||||||
| > > | It is generally preferred to isolate genomic DNA from your culture before doing the 16s RNA PCR (using the Invitrogen PureLink kit, for example). This results in a cleaner PCR product and generally a better sequencing result. | ||||||||
| Changed: | |||||||||
| < < | Use the invitrogen purelink genomic kit to isolate DNA from your culture. | ||||||||
| > > | After DNA is isolated, use a Qubit to find the concentration of DNA. The desired amount of gDNA template for PCR is 0.1 ng/ul (final template concentration) so for a 30 ul PCR reaction use 3 ng of DNA. | ||||||||
| Deleted: | |||||||||
| < < | After DNA is isolated, use a nanodrop to find the concentration of DNA. The ideal amount of DNA for PCR is 0.1 ng/ul (final template concentration) so for a 30ul PCR reaction use 3ng of DNA. | ||||||||
The PCR cycle is as follows:
| |||||||||
| Changed: | |||||||||
| < < | After PCR, the products need to be run on an agarose gel to confirm correct product (around 1kb) and to excise the correct bands from the gel. | ||||||||
| > > | After PCR, the products need to be run on an agarose gel to confirm correct product (around 1kb). A 1.5% or 2% gel works the best for this. | ||||||||
| Changed: | |||||||||
| < < | A 1.5% - 2% gel works the best for this. | ||||||||
| > > | If the bands are clean, then you can do a PCR cleanup spin column and submit the DNA for two different sequencing reactions (one with each of your PCR primers). If the bands are not clean, you may be able to excise the band and | ||||||||
| Deleted: | |||||||||
| < < | After cutting the bands out of gel clean using Zymo gel extraction kit or equivalent. To prepare DNA for sequencing follow the instructions found here: http://www.icmb.utexas.edu/core/DNA/DNA_Sequencing/FAQ-Sequencing.htm | ||||||||
| Added: | |||||||||
| > > | The bands should be clean to excise the correct bands from the gel. | ||||||||
| Added: | |||||||||
| > > | After cutting the bands out of gel clean using Zymo gel extraction kit or equivalent. To prepare DNA for sequencing follow the instructions found here: http://www.icmb.utexas.edu/core/DNA/DNA_Sequencing/FAQ-Sequencing.htm | ||||||||
Analysis | |||||||||
| Changed: | |||||||||
| < < | Use the tools at the Ribosomal Protein Database | ||||||||
| > > | Use the Classifer tool | ||||||||
References1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003). | |||||||||
Revision 52011-07-15 - CraigBarnhart
16S rRNA Sequencing to Identify Unknown Microbes | |||||||||
| Changed: | |||||||||
| < < | Ever wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate? | ||||||||
| > > | Ever wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate? | ||||||||
| Added: | |||||||||
| > > | Overview
| ||||||||
PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
| |||||||||
| Added: | |||||||||
| > > |
It is generally preferred to isolate the DNA from your culture before doing the 16s RNA PCR. This results in a cleaner PCR product and generally a better sequencing result. Use the invitrogen purelink genomic kit to isolate DNA from your culture. After DNA is isolated, use a nanodrop to find the concentration of DNA. The ideal amount of DNA for PCR is 0.1 ng/ul (final template concentration) so for a 30ul PCR reaction use 3ng of DNA. The PCR cycle is as follows:
After PCR, the products need to be run on an agarose gel to confirm correct product (around 1kb) and to excise the correct bands from the gel. A 1.5% - 2% gel works the best for this. After cutting the bands out of gel clean using Zymo gel extraction kit or equivalent. To prepare DNA for sequencing follow the instructions found here: http://www.icmb.utexas.edu/core/DNA/DNA_Sequencing/FAQ-Sequencing.htm | ||||||||
AnalysisUse the tools at the Ribosomal Protein DatabaseReferences1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003). | |||||||||
Revision 42011-06-07 - CraigBarnhart
16S rRNA Sequencing to Identify Unknown MicrobesEver wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
| |||||||||
| Changed: | |||||||||
| < < | The optimal dilution is somewhere between 1000 and 10,000 fold. PCR setup should include an initial denaturing step: 10min at 94 C. | ||||||||
| > > | The optimal dilution is somewhere between 1000 and 10,000 fold. PCR setup should include an initial denaturing step: 10min at 94 C. | ||||||||
AnalysisUse the tools at the Ribosomal Protein DatabaseReferences1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003). | |||||||||
Revision 32011-04-25 - CraigBarnhart
16S rRNA Sequencing to Identify Unknown MicrobesEver wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
| |||||||||
| Added: | |||||||||
| > > | The optimal dilution is somewhere between 1000 and 10,000 fold. PCR setup should include an initial denaturing step: 10min at 94 C. | ||||||||
AnalysisUse the tools at the Ribosomal Protein DatabaseReferences1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003). | |||||||||
Revision 22010-05-26 - JeffreyBarrick
16S rRNA Sequencing to Identify Unknown MicrobesEver wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
| |||||||||
| Added: | |||||||||
| > > | The dilution of cells used in the reaction plays a critical role in the success of the amplification. Too many cells or components of certain media can inhibit PCR. For best results take a visible turbid overnight culture from a rich medium and dilute approximately 10,000-fold into the final PCR. Ideally, make a dilution series of the template cells. | ||||||||
AnalysisUse the tools at the Ribosomal Protein DatabaseReferences1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003). | |||||||||
Revision 12010-05-13 - JeffreyBarrick
16S rRNA Sequencing to Identify Unknown MicrobesEver wonder what that contaminant in your culture is? Need to accurately identify an environmental isolate?PCR ReactionFor PCR we use universal primers U341F and UA1406R that amplify an approximately 1 kb stretch of rDNA and should work for nearly any bacterial or archaeal sequence [1].
AnalysisUse the tools at the Ribosomal Protein DatabaseReferences1. Baker, G.C., Smith, J.J. & Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55, 541-55 (2003). |
View topic | History: r9 < r8 < r7 < r6 | More topic actions...
Ideas, requests, problems regarding TWiki? Send feedback